#include <iostream>#include <fstream>#include <string>#include <cmath>#include <assert.h>#include <cstdlib>#include <limits>#include <cuda.h>#include <ctime>#include "gpu_forces.cuh"#include "gpu_integrator.cuh"#include "gpu_tools.cuh"#include "define.h"#include "data.h"#include <cstring>#include "tests.cuh"Go to the source code of this file.
Functions | |
| int | main (int argc, char *argv[]) |
| Main function. | |
mdgpu - a molecular dynamic simulation program using GPUs
Copyright (C) 2010 Julian P. Engel
This program is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with this program. If not, see <http://www.gnu.org/licenses/>.
Definition in file main.cu.
| int main | ( | int | argc, | |
| char * | argv[] | |||
| ) |
Used to build up the molecular dynamics simulation loop.
| [in] | argc | Number of command line arguments. Have to be 14 and in the following order. In case of unsufficient passed arguments the program will stop. | ||||||||||||||||||||||||||||
| [in] | argv | This array holds the 14 command line arguments:
|
Build in fixed parameters:
rCut | cutoff radius of the Lennard-Jones potential set to |
rVer | cutoff radius for the Verlet list of each particle: |
numPointsPerDecade | Number of points per decade in logarithmic output. |
threads | Maxium number of threads per thread block on the GPU is set to |
redM | Number of values after first level of reduction is set to |
redN | Number of threads for second reduction level set to |
Definition at line 64 of file main.cu.
References checkCudaError(), FLOAT_ARRAY_TYPE, generateStartScript(), gpuGenerateVerletListSmem(), loadInitData(), memInfo(), removeDrift(), rescaleT(), setCudaDevice(), and writeData().
 1.6.1
1.6.1
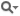

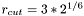
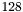 (could be up to 512).
(could be up to 512). 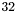 to occupy full blocks in the second reduction level.
to occupy full blocks in the second reduction level.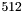 ).
).