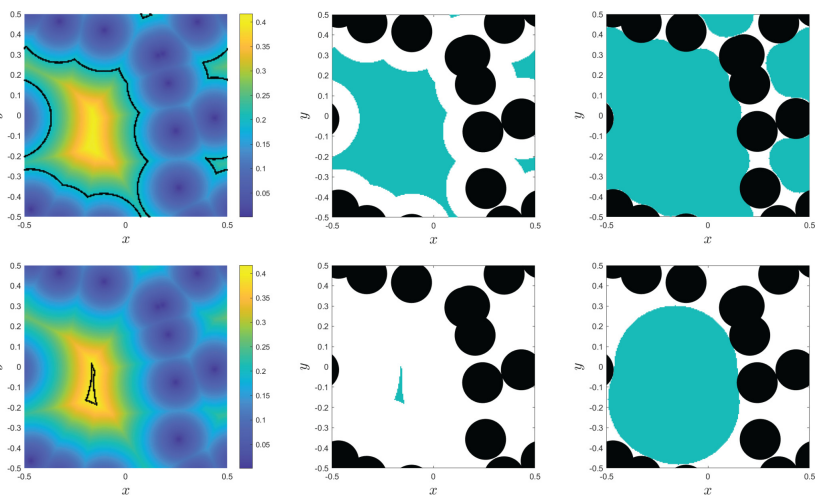
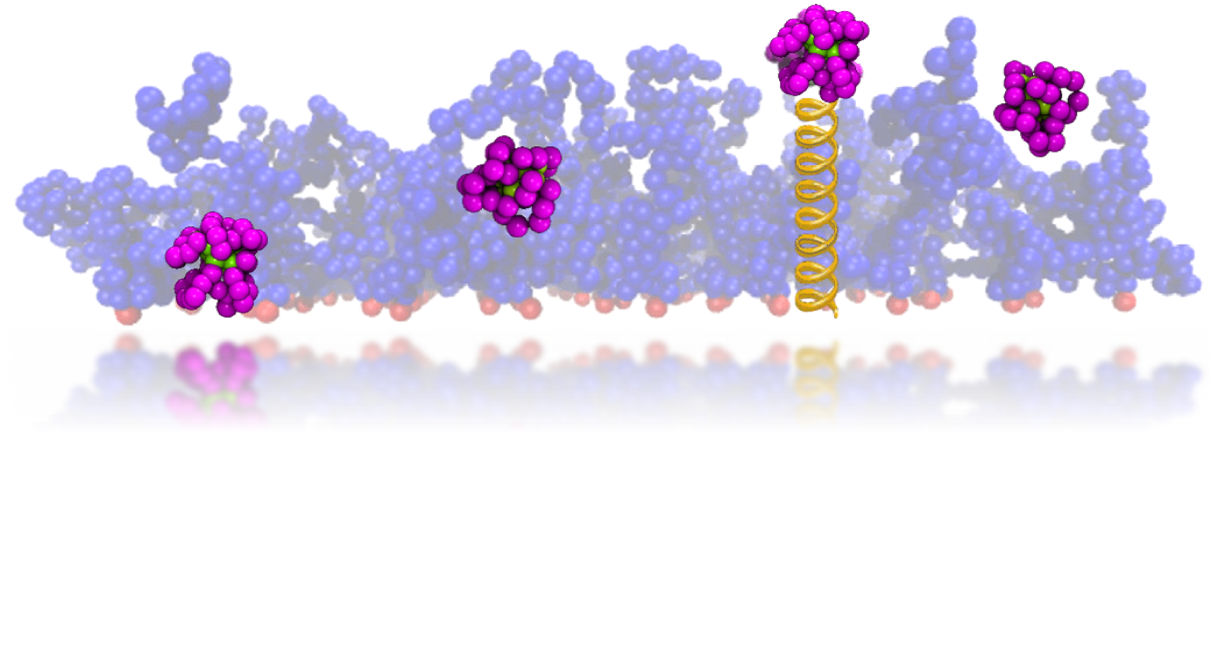
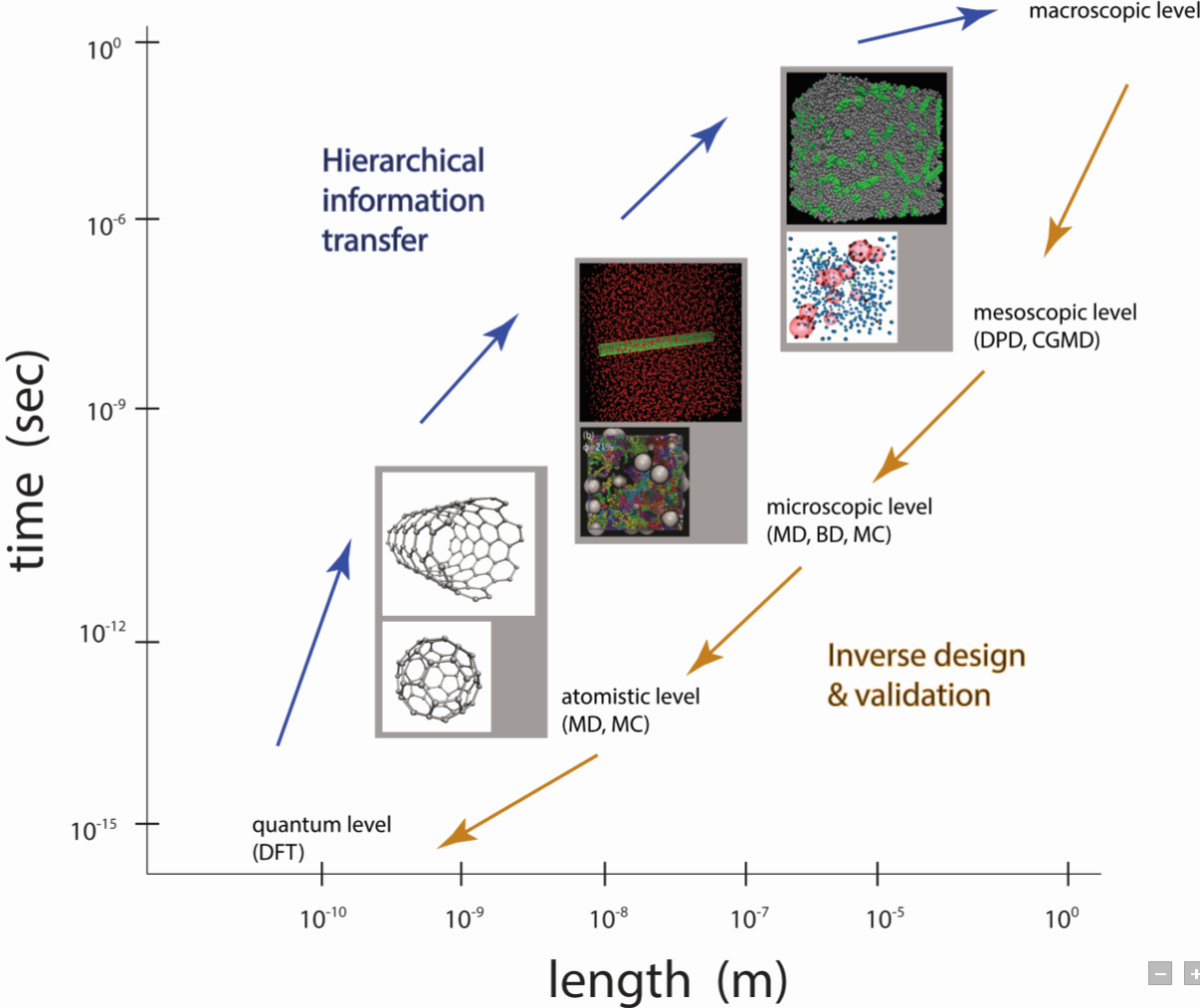
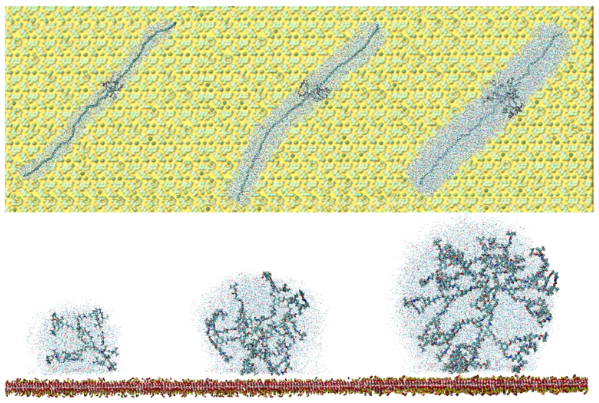
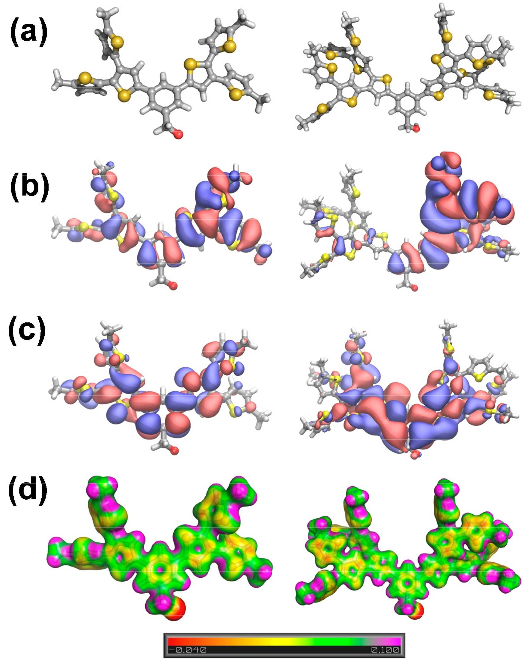
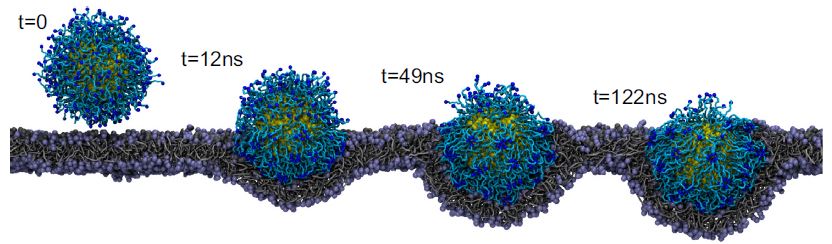
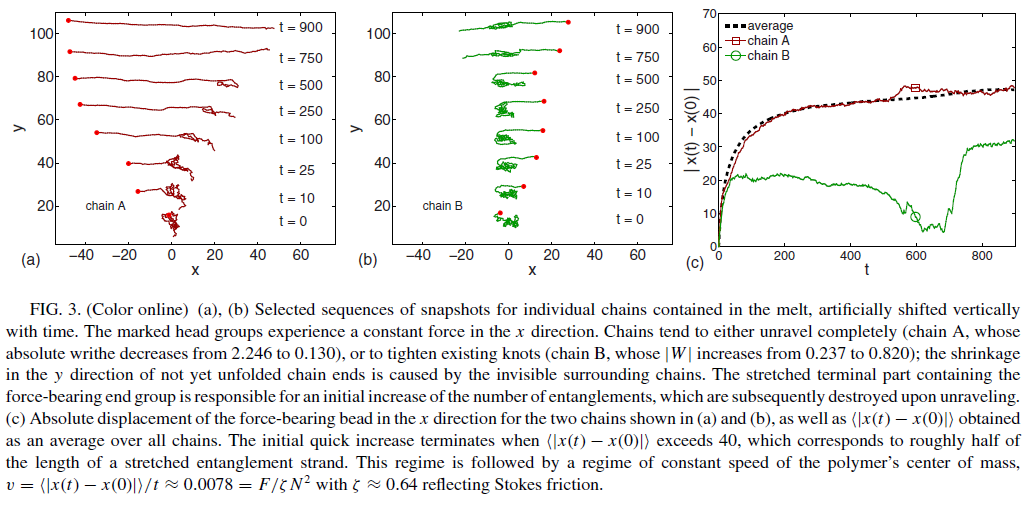
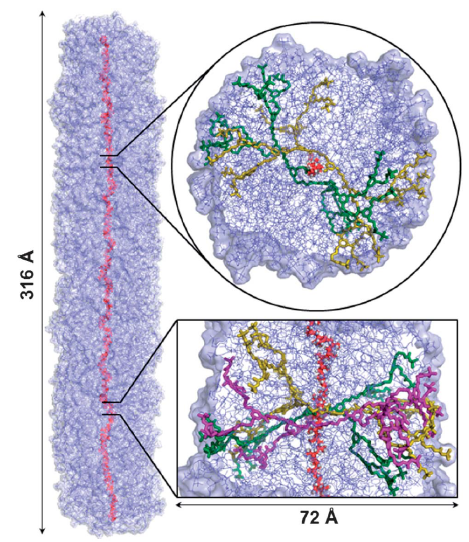
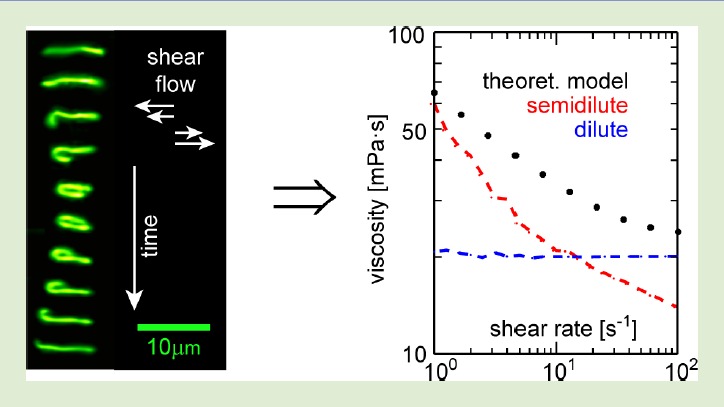
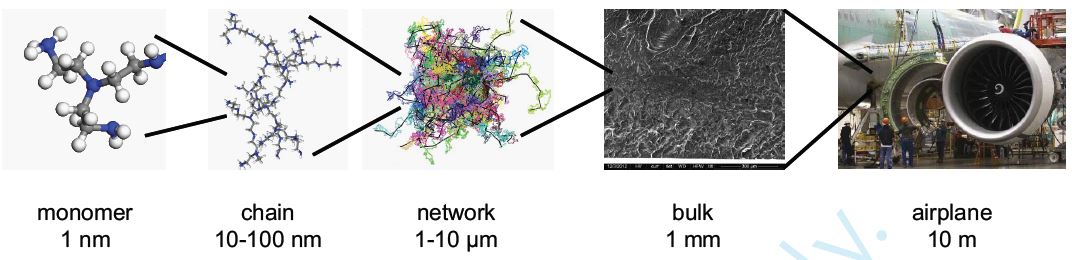
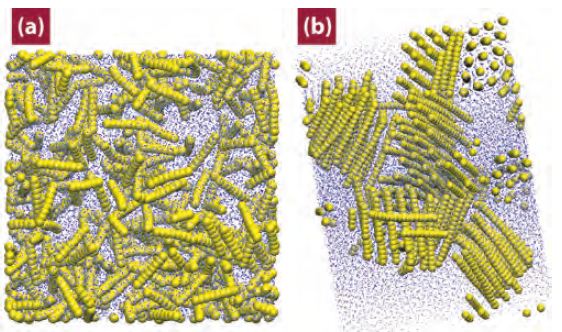
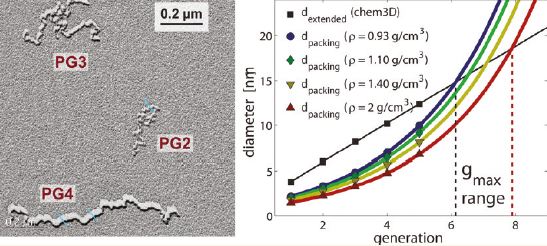
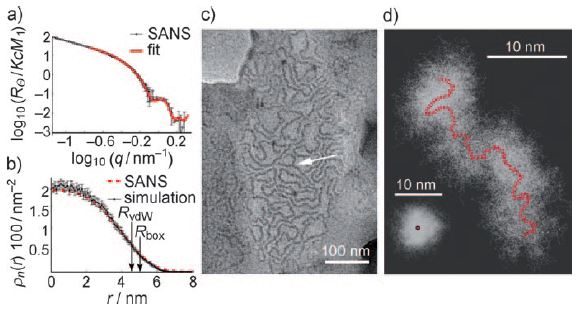
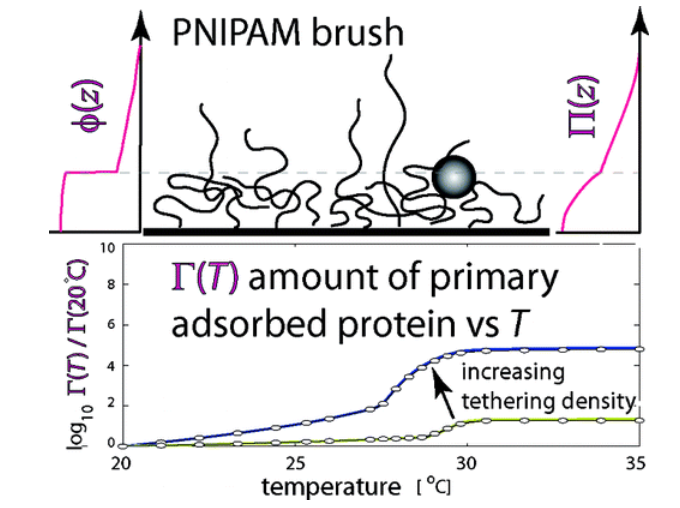
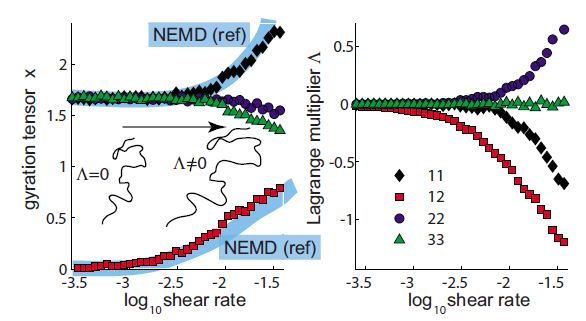
The size, shape, surface property and material composition of polymer-coated nanoparticles (NPs) are four important parameters in designing efficient NP-based carriers for targeted drug delivery. However, due to the complex interplay between size, shape and surface property, most studies lead to ambiguous descriptions of the relevance of shape. To clarify its influence on the cellular uptake of PEGylated NPs, large scale molecular simulations have been performed to study differently shaped convex NPs, such as sphere, rod, cube and disk. Comparing systems with identical NP surface area, ligand.receptor interaction strength, and grafting density of the polyethylene glycol, we find that the spherical NPs exhibit the fastest internalization rate, followed by the cubic NPs, then rod- and disk-like NPs. The spherical NPs thus demonstrate the highest uptake among these differently shaped NPs. Based on a detailed free energy analysis, the NP shape effect is found to be mainly induced by the different membrane bending energies during endocytosis. The spherical NPs need to overcome a minimal membrane bending energy barrier, compared with the non-spherical counterparts, while the internalization of disk-like NPs involves a strong membrane deformation, responsible for a large free energy barrier. Besides, the free energy change per tethered chain is about a single kBT regardless of NP shape, as revealed by our self-consistent field theory calculations, where kB and T denote Boltzmann constant and temperature, respectively. Thus, the NP shape only plays the secondary role in the free energy change of grafted PEG polymers during internalization. We also find that star-shaped NPs can be quickly wrapped by the cell membrane, similar to their spherical counterparts, indicating star-shaped NPs can be used for drug delivery with high efficacy. Our findings seem to provide useful guidance in the molecular design of PEGylated NPs for controllable cellular uptake and help establish quantitatively rules in designing NP-based vectors for targeted drug delivery. |

homepage 293 out of 848 entries requested